
About Our Lab:
In response to specific stimuli, cells, through a coordinated action of many proteins forming a so-called signal transduction cascade or network, will generate a specific response. Achieving an understanding of these signaling networks in tissue culture models let alone in living individuals is a major challenge. The complexity and dynamic behavior of individual signaling cascades is such that the simple knowledge of the players (proteins) and their interacting partners is not enough to provide an accurate quantitative description of the system. Molecular Imaging provides for a unique opportunity to quantify these signaling cascades in a non-invasive and dynamic manner. The development of genetically encoded proteins, which enable real-time, high-resolution and semi-quantitative visualization of molecular concentrations and protein-protein interactions. More interestingly, the ability to image specific enzymatic activities (kinase, phosphatase, proteases, glycanase etc.) within a signaling cascade dynamically and quantitatively will serve as key experimental tools that will ultimately allow an intuitive understanding of the signal–response relation. The Rehemtulla laboratory has used the ability image signaling cascades in mouse models to provide novel insights into the biology of cancer. Additionally, cell based have been used to delineate novel signaling events that promote the transformed phenotype.
Projects
Regulation of TGFβ signaling
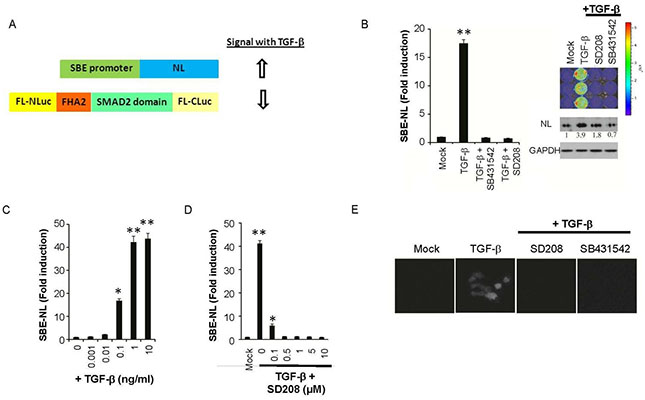
The transforming growth factor-β (TGFβ) family of cytokines regulates many processes such as immune suppression, angiogenesis, wound healing, and epithelial to mesenchymal transition (EMT). Abnormal TGFβ signaling is linked to autoimmune and autoinflammatory diseases, fibrosis, tumor formation and metastasis, and various other disorders. Early in tumorigenesis, the proliferation of epithelial cells retains exquisite sensitivity to TGFβ, wherein TGFβ elicits a tumor suppressive response. The binding of TGFβ to TGFβ receptor-2 (TGFBRII) induces the interaction between TGFβ receptor-1 (TGFBRI) and TGFBRII leading to the phosphorylation and activation of transcriptional regulators SMAD2 and SMAD3. An siRNA screen of the human kinome using a live-cell reporter for TGFBR kinase activity identified budding uninhibited by benzimidazoles-1 (BUB1), a Ser/Thr kinase, as an essential mediator of TGFβ signaling. BUB1 interacted with TGFBRI in response to stimulation with TGFβ and promoted its interaction with TGFBRII. A small-molecule inhibitor of BUB1 kinase (2OH-BNPP1) and a kinase-deficient mutant of BUB1 abrogated ligand-mediated canonical and non-canonical TGFβ signaling in various cancer and normal cell lines. 2OH-BNPP1 administration to mice bearing A549 xenografts reduced the amount of phosphorylated SMAD2 in tumor tissue. These findings provide evidence of a role of BUB1 as a kinase in mediating TGFβ dependent signaling beyond its established function in cell-cycle regulation and chromosome cohesion.
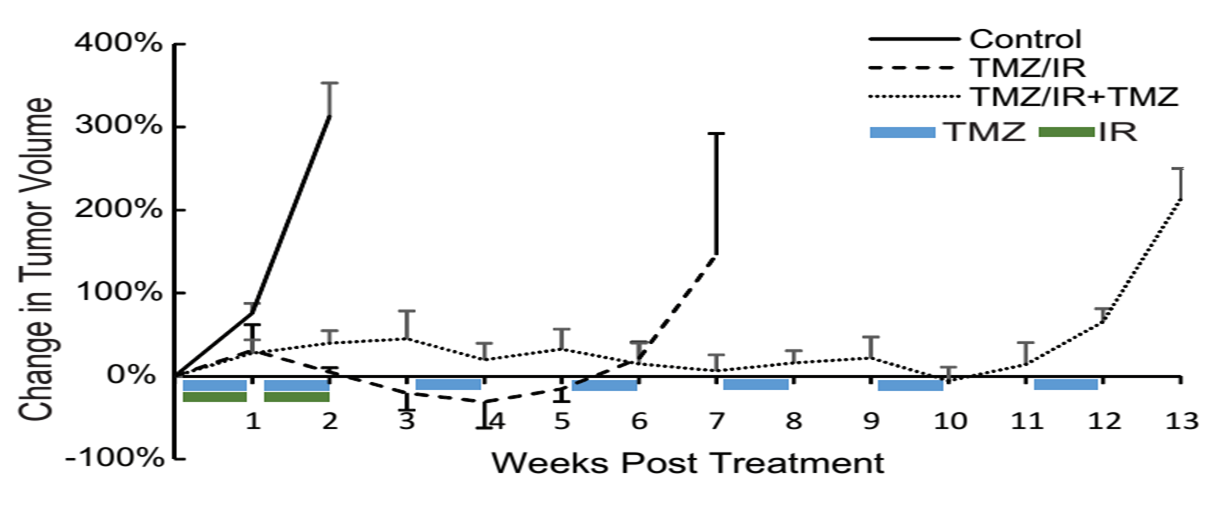
Genomics reveals a role for TGFβ signaling in therapeutic resistance in glioblastoma
Glioblastoma multiforme (GBM) represents the most aggressive malignant primary brain tumor. Radiation (IR) with concomitant and adjuvant temozolomide (TMZ) after surgical resection, is the standard of care for newly diagnosed GBM patients. Despite an initial response, most patients succumb to the disease due to the development of resistance such that the median survival is only 15 months. In an effort to delineate the mechanistic basis for the evolution of resistance to TMZ and IR, we established intracranial gliomas in mice, using patient derived tumor cells. Samples were collected pre-treatment and post-recurrence using MRI-guided biopsy and analyzed using whole genome RNA sequencing and validated using biochemical studies. Gene expression profiling of pre-treatment and TMZ/IR resistant tumors revealed an upregulation of a network of genes within the mesenchymal and stem cell signatures and downregulation of genes involved in cell death. Based on the role TGF-β in regulating mesenchymal transition and imparting tumor cell self-renewal capacity in GBM, we evaluated if inhibition of this pathway restored the sensitivity of recurrent tumors to TMZ/IR. Our studies show that upregulation of mesenchymal and stem cell genes by the TGF-β signaling pathway contributes to therapeutic resistance, and provides a rationale for clinical trials wherein TGF-β inhibitors can be used to prevent or reverse the therapy resistant phenotype commonly observed in GBM.
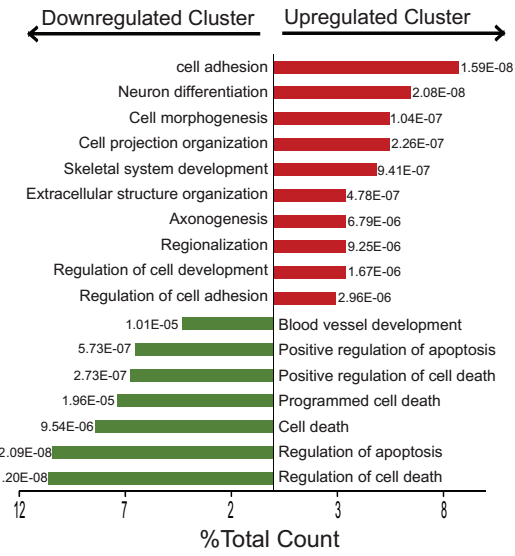
Identification of FADD as a key mediator of KRAS oncogenic activity
Dysregulated cell signaling and proliferation occurs through overexpression, post-translational modification or mutation of signaling proteins. RAS is a membrane associated small G protein that functions as a signaling mediator of receptor and non-receptor tyrosine kinases to cytoplasmic and nuclear effector pathways. Oncogenic mutations of RAS, account for approximately 30% of all cancers, which results in constitutive signaling, leading to dysregulated cell proliferation and enhanced survival. Mutations within the KRAS gene are common in non-small-cell lung cancer (NSCLC), colorectal, and pancreatic cancer. Although genomic amplification and phosphorylation of Fas Associated Death Domain (FADD) has been associated with poor clinical outcome in lung and head and neck cancer, its role in oncogenesis is not fully understood. Using conditional mouse models of Kras-driven lung cancer, we demonstrate a requirement for FADD in lung cancer initiation and progression. In the absence of FADD, abrogation of tumor growth was observed wherein a lower proliferative index and decreased activation of downstream effectors of the RAS-mitogen-activated protein kinase (MAPK) pathway, including phosphorylated extracellular signal-regulated kinase 1 and 2 (pERK1/2), phosphorylated retinoblastoma (pRB) and CyclinD1, indicated alterations in cell cycle progression. Studies using embryonic fibroblasts revealed that the induction of mitogenesis upon activation of the RAS-MAPK pathway required FADD and its phosphorylation by Casein Kinase 1 alpha (CK1α) A conditional mouse wherein CK1α expression was ablated simultaneous with Kras activation, confirmed a requirement for FADD-phosphorylation in Kras-mediated lung oncogenesis.
Join Us:
Principal Investigator

Lab members
