Section by Dr. Eva Feldman:
PATHOPHYSIOLOGY OF DPN
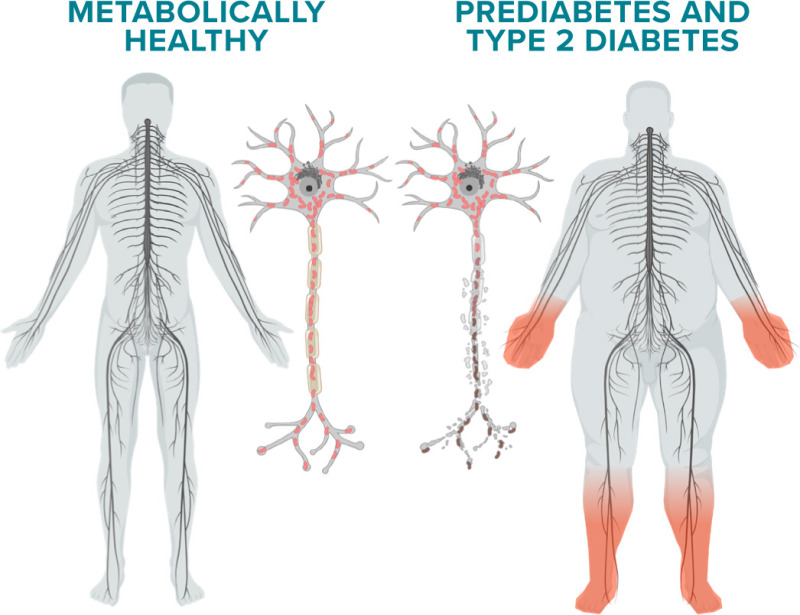
Diabetes preferentially affects the peripheral nervous system (PNS), a likely reflection of the unique anatomy of the PNS (21). PNS axons are frequently ≥3 feet long and >20,000 times the length of their supporting cell bodies. PNS sensory neurons and their receptors lie outside the blood-brain barrier and are more vulnerable to injury secondary to diabetes than motor neurons, which lie within the barrier. Among the sensory neurons, there are small unmyelinated neurons known as C-fibers but also frequently called “small fibers.” These fibers carry nociceptive information, particularly related to heat and pain, and constitute the majority of sensory axons in the PNS. The lack of myelin results in slow, continuous conduction of small fibers secondary to a uniform distribution of ion channels along the axon. In conjunction with small fibers are small, thinly myelinated Aδ fibers, which relay information on touch, pressure, and cold, and fully myelinated fibers of different diameters, designated Aβ and Aα, which are responsible for vibratory and position sense. Collectively, these fiber types are known as large fibers. Myelin, provided by Schwann cells, ensheathes the axons of these fibers in a highly controlled manner, forming the nodes of Ranvier and paranodes, the sites of ion channels required for rapid nerve conduction and of tight junctions that protect large fibers from toxic substances (21).
Anatomical studies from sural nerve biopsies of patients with diabetic neuropathy align with their presenting symptoms (22). Early degeneration and loss of C fibers are evident in patients experiencing new-onset pain, burning, or prickling, which are known as dysesthesias, in their feet, followed by initial demyelination/remyelination of large fibers. As the disease progresses, large fiber axonal loss eventually occurs, and patients experience numbness and loss of proprioception in the feet that travel upward over time. This distal-to-proximal axonal loss and its accompanying symptoms are the hallmarks of diabetic neuropathy (23).
Between 1970 to 2010, studies aiming to understand the pathophysiology of diabetic neuropathy focused on glucose dysregulation (24). In the polyol pathway, aldose reductase converts excess glucose to sorbitol, resulting in a series of downstream reactions that decrease sodium-potassium adenosine triphosphatase (ATP) activity, deplete nicotinamide adenine dinucleotide phosphate, and produce reactive oxygen species (ROS), impairing nerve function. Aldose reductase inhibitors were tested in 32 diabetic neuropathy clinical trials but unfortunately failed to improve nerve function.
Excess glucose also enters the hexosamine pathway, producing inflammatory by-products and activating protein kinase C (PKC) secondary to the accumulation of diacylglycerol. PKC activation, in turn, enhances insulin resistance, disrupts growth factor biology, and leads to vasoconstriction of nerve blood vessels. Similar to the aldose reductase trials, clinical trials of PKC inhibitors failed in human diabetic neuropathy.
Advanced glycation end products (AGEs), which bind receptors for AGEs (RAGEs), are another byproduct of excess glucose. Activation of AGEs and RAGEs leads to downstream inflammation, ROS accumulation, and decreased blood flow to peripheral nerves. Although preclinical trials targeting activation of RAGEs were promising, available compounds were too toxic for human trials and remained in therapeutic development (25).
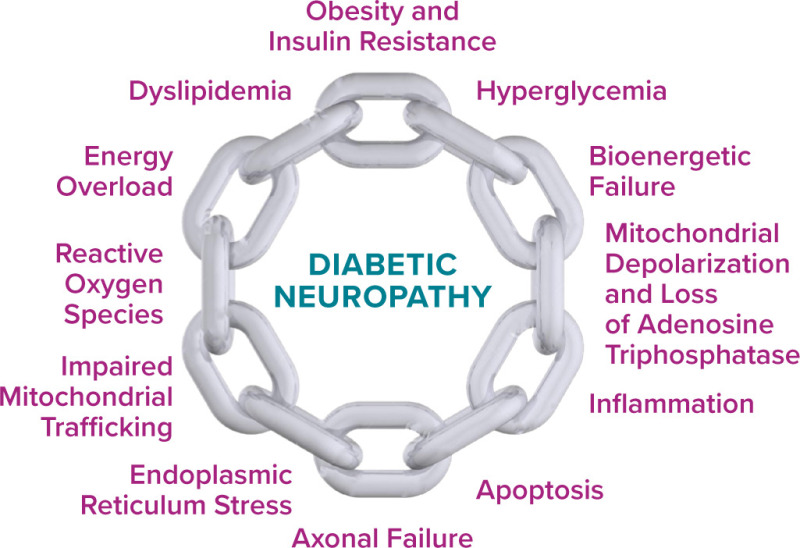
Many of the failed clinical trials occurred at the same time multiple, newer clinical trials were suggesting that glucose control alone was insufficient to prevent neuropathy in people with type 2 diabetes (26). There is now consensus that glycemic control alone cannot prevent the progression of DPN in patients with type 2 diabetes. The metabolic syndrome has emerged as a crucial risk factor for neuropathy based on data from multiple clinical studies in the United States (4,27–29), Denmark (15), Germany (30), the Netherlands (31), India (32), and China (33,34). The metabolic syndrome encompasses hyperglycemia, obesity, and dyslipidemia, and the risk of developing neuropathy increases with the number of these components present in an individual (27,28). These clinical trials led to new thinking about the pathophysiology of diabetic neuropathy focused on the idea that disruption in whole-nerve bioenergetics (i.e., how the nerve accesses energy along its entire length) is the crucial factor leading to disease (35).
Mitochondria are the energy-producing organelles in cells and use both glucose and lipids to produce ATP. In the PNS, mitochondria are primarily made in the cell body and are trafficked down long axons to provide energy for nerve function (36). In small fibers, mitochondria are found along the length of the axons. In large fibers, they are also present along the length of the axons but are particularly present at the nodes of Ranvier and the paranodes, where they assist in salutatory nerve conduction. In both fiber types, mitochondria produce ATP from glucose and lipids (21). ATP then supplies the needed energy for nerve impulses to travel the length of the axons and reach distal nerve terminals.
Under normal conditions, glucose and lipids undergo a series of distinct, highly regulated chemical reactions, ending with the transfer of the electron donors nicotinamide adenine dinucleotide and flavin adenine dinucleotide to the mitochondrial electron transport chain. These electron donors travel across the inner mitochondrial membrane and undergo oxidative phosphorylation, with the generation of ATP for energy and small amounts of ROS as a by-product of the process. However, in the diabetic environment, excess glucose and lipids not only disrupt the normal pathways used for their own breakdown but also produce excess electron donors that the mitochondria are unable to process. The result is a bioenergetic failure (37) and the loss of normal mitochondrial membrane function (mitochondrial depolarization), decreased ATP production, impaired mitochondrial trafficking, and accumulation of ROS, leading to inflammation, endoplasmic reticulum stress, apoptosis of neurons, and axonal failure (38) (Figure 1).